A Predictive Model for Distant Metastasis in Patients With Breast Cancer Based on Machine Learning
Article information
Abstract
Purpose
Breast cancer starts as a local disease, but can metastasize to distant organs. In this study, we described an easy-to-use tool for predicting distant metastases based on clinical characteristics and gene expression profiles.
Methods
We performed a retrospective chart review of 326 patients with breast cancer who underwent surgery and CancerSCANTM between January 2001 and December 2014 at the Samsung Medical Center. Additional retrospective data for 83 patients during 2015 were used for internal validation. CancerSCANTM, a next-generation sequencing-based targeted deep sequencing analysis, was used for gene analysis, and Azure Machine Learning (ML) was used for the ML processes.
Results
The no-distant metastasis group comprised 267 patients, while the distant metastasis group comprised 59. Using the Azure ML platform, a predictive model was developed with 326 cases. The area under the curve of the receiver operating characteristic curve for predictive value was 0.917. Based on the internal validation performed using 83 patients, the true-negative was 81 and the true-positive was two when a threshold value of 0.5 was applied.
Conclusion
Patients with breast cancer are at risk of metastasis and experience fear throughout their lives. Our predictive model is a valuable and easy-to-access tool for identifying patients with distant metastasis and it presents a way for each institution to achieve optimal results using its variables. Further evaluation with a larger patient population will improve the reliability of this model.
INTRODUCTION
Breast cancer is the most common cancer among women worldwide [1]. Breast cancer starts as a local disease but can metastasize to the lymph nodes and distant organs [2]. Despite advances in breast cancer therapy, 20%−30% of patients with early breast cancer experience relapse with distant metastatic disease [3]. Tumor metastasis is a major clinical challenge for most cancer-related deaths [4].
In previous studies, the prediction of distant metastasis was based on intrinsic biological subtypes [5,6] and clinical status, including tumor size and nodal status [7]. Gene expression profiles have been used as predictive markers for distant metastasis [8,9]; however, clinical application has been difficult.
In this study, we present an easy-to-use tool for predicting distant metastases based on clinical characteristics and gene expression profiles. Gene profiles were obtained from CancerSCANTM, a targeted sequencing platform designed at the Samsung Medical Center [10].
METHODS
Study population
We performed a retrospective chart review of 336 patients with breast cancer who underwent surgery and CancerSCANTM between January 2001 and December 2014 at the Samsung Medical Center in Seoul, Korea. DNA sequencing results and electronic medical records, including pathology reports, were reviewed. Ten cases were excluded from the analysis owing to incomplete medical data. As a result, 326 cases were included in the analysis. For internal validation, additional retrospective data from 83 patients who underwent surgery and CancerSCANTM in 2015 were used. This study adhered to the tenets of the Declaration of Helsinki and was approved by the Institutional Review Board (IRB) of Samsung Medical Center (IRB no. 2018-05-005).
The available data for the cohorts included age at diagnosis, subtype (e.g., hormone receptor [HR]+/human epidermal growth factor receptor 2 [HER2]-, HR+/HER2+, HR-/HER2+, HR-/HER2-), histopathology (e.g., invasive ductal carcinoma [IDC], other), operation type (e.g., breast-conserving surgery, total mastectomy, sentinel lymph node biopsy, axillary lymph node dissection), chemotherapy (e.g., neo-adjuvant, adjuvant, none), regimen (e.g., adriamycin, cyclophosphamide [AC]), AC+docetaxel/taxol, fluorouracil, adriamycin, Cytoxan, AC, methotrexate, fluorouracil, docetaxel, carboplatin, trastuzumab, pertuzumab, and others, radiotherapy, hormonal therapy, target therapy, nuclear grade, pathological T-stage, pathological N-stage, distant metastasis, and metastatic site. Distant metastasis was defined as distant detectable metastasis confirmed using clinical and radiographic means and histologically proven for lung, liver, and peritoneal metastases. For bone and brain metastases, definite radiological findings were considered distant metastasis without biopsy. Positron emission tomography, chest computed tomography (CT), abdominal CT, bone scans, and brain magnetic resonance imaging were used to obtain radiological findings.
Targeted deep sequencing using a customized cancer panel (CancerSCAN™)
Genomic DNA (250 ng) from each tissue sample was sheared in a Covaris S220 ultrasonicator (Covaris, Woburn, USA) and used with CancerSCANTM probes and the SureSelect XT reagent kit, HSQ (Agilent Technologies, Santa Clara, USA), to construct a library, according to the manufacturer’s protocol.
The panel was designed to enrich the exons of 81 genes, covering 366.2 kb of the human genome. After multiplexing, the enriched exome libraries were sequenced on a HiSeq 2500 sequencing platform (Illumina). A paired-end DNA sequencing library was prepared via gDNA shearing, end repair, A-tailing, paired-end adaptor ligation, and amplification. After hybridization of the library with bait sequences for 27 hours, the captured library was purified and amplified using an index barcode tag, and the library quality and quantity were assessed.
The exome library was sequenced using the 100-bp paired-end mode of the TruSeq Rapid PE Cluster Kit and TruSeq Rapid SBS Kit (Illumina).
Sequence reads were mapped to the human genome (hg19) using the Burrows-Wheeler Aligner [11]. Duplicate reads were removed using the Picard and SAM tools [12]. Local alignments were optimized using the Genome Analysis Toolkit [13]. Variant calling was only performed in regions targeted by CancerSCANTM. To detect single nucleotide variants, we integrated the results of the three types of variant callers, which increased the sensitivity [14]. A Pindel was used to detect the indels [15]. Copy number variations were calculated for the targeted regions by dividing the read depth per exon by the estimated normal reads per exon using an in-house reference.
Gene profiles
CancerSCANTM is a next-generation targeted deep sequencing analysis method covering 81 genes. Gradient boosting was performed to identify the genes important for survival. In particular, the input features were alterations in 81 genes, including loss-of-function, mutation, and copy number variations. The target value was alive or dead at the five-year mark. Important features were identified by serially reducing their number of features [16]. The hyperparameters were optimized as follows: number of estimators, 999; learning rate, 0.15; and maximum depth, 6. Bootstrap resampling (n = 100) was performed in which the training sets (85%) and their corresponding test sets (15%) were resampled 100 times to evaluate the internal stability of the model. Wilcoxon test was used to determine the optimal number of genes (Supplementary Figure 1) [17].
Statistical analysis
Variables were compared between the no-distant and distant metastasis groups using the chi-square or Fisher’s exact test. Mean age was compared between the two groups using the Mann-Whitney U test with SAS version 9.4 (SAS Institute, Cary, USA). Receiver operating characteristic (ROC) curves and areas under the ROC curves (AUCs) were calculated. All tests were two-sided, and a p < 0.05 was considered to indicate statistical significance.
Machine learning (ML)
Azure ML (Microsoft, Redmond, USA) is a cloud service that enables the execution of ML processes. The Azure ML Studio (Microsoft, Redmond, USA) is available as a workspace to help users build and test predictive models [18]. We built a supervised ML classification model using the Azure ML platform by performing the following: (1) data editing, (2) data splitting, (3) model training, (4) model scoring, and (5) model evaluation (Figure 1). We split the modeling data (326 cases) into training and testing sets using a randomized 50–50 split. Thereafter, we trained our training set using the Two-class Decision Forest method [19] to predict distant metastasis and the Multi-class Neural Network [20] method to predict distant metastatic sites.

Modeling workflow using Azure Machine Learning. The workflow included establishing a dataset (e.g., 27 gene_azureml), editing the metadata, employing an algorithm (e.g., two-Class Decision Forest), splitting the data, training the model, scoring the model, and evaluating the model.
RESULTS
Patient characteristics
Table 1 shows the baseline characteristics of patients. The no-distant metastasis group comprised 267 patients, and the distant metastasis group comprised 59. The median follow-up period was 104 months (range, 1–203) and the average distant metastasis-free interval was 85 months (range, 1–190). HR+/HER2- was higher in the no-distant metastasis group (p = 0.011), while HR-/HER2+ was higher in the distant metastasis group (p = 0.003). The distant metastasis group had more patients 50 years and older (p = 0.000). Further, the mean age was slightly higher in the distant metastasis group than in the no-distant metastasis group; however, the difference was not significant. IDC accounted for the majority in both groups, with no difference found according to the surgical type. More patients received neoadjuvant chemotherapy and radiotherapy in the distant metastasis group than in the no-distant metastasis group; opposite results were obtained for adjuvant chemotherapy and hormonal therapy. Nuclear grades 1 and 2 and pathological T stage did not differ between the two groups; however, nuclear grade 3, nodal stage, and pathological stage were found to differ between the two groups. The no-distant metastasis group had higher N0 (p = 0.000), while the distant metastasis group had higher nuclear grade 3, and N2 and 3 (p = 0.010, p = 0.000, p = 0.001, respectively). Regarding the pathological stage after surgery, the no-distant metastasis group had a higher number of stage II cases (p = 0.003), while the distant metastasis group had a higher number of stage III cases (p = 0.000).

The baseline characteristics of enrolled patients (n=326)
Distant metastasis
Table 2 shows the proportion of metastatic sites in the distant metastasis group. Among the 59 patients, 21 (35.6%) had multiple metastatic sites and 19 (32.2%) had lung metastases. Only one other site of metastasis, the contralateral supraclavicular lymph node, was found. of the 21 cases of multiple-site metastasis, six were triple-site cases (28.6%) and 15 were double-site cases (71.4%).

Metastatic sites in distant metastasis group (n=59)
Among the multiple-site metastasis cases, the cumulative counts for lung, bone, liver, brain metastasis, and peritoneal seeding were 17, 12, 10, 7, and 2, respectively.
Gene signature
We used the results of 27 genes and 34 occasions from the CancerSCANTM data for the analysis. Table 3 shows the cumulative counts of each gene signature between the two groups. PIK3CA mutations were the most frequent gene variations among patients. 34.5% of the no-distant metastasis group and 27.1% of the distant metastasis group had PIK3CA mutation. BRCA1 loss-of-function and BRCA2 were more frequent in the distant metastasis group than in the no-distant metastasis group; however, the total counts were very small. Of the 59 patients with distant metastasis, 1 (1.7%) had seven gene variations and 6 (3.4%) had no gene variation. Most patients (16, 27.1%) in the distant metastasis group had two genetic variations. Among the 267 patients without distant metastasis, 1 (0.4%) had 10 gene variations, and 28 (10.5%) had no gene variation. Most patients (81, 30.3%) in the no-distant metastasis group had two genetic variations.

The differences of gene signature between no distant metastasis group and distant metastasis group
Predictive model
We developed a predictive model with 326 cases using the Azure MLplatform (Figure 1) with various classification algorithms, such as two-class Decision Forest, two-class Decision Jungle, two-class Bayes Point Machine, two-class Support Vector Machine, and two-class Neural Network. Of the algorithms, the two-class Decision Forest method was identified as the most suitable for predicting distant metastasis. Based on the calculations, the AUC was 0.917 and the accuracy was 0.903 (Figure 2). Internal validation was conducted using 83 patients who underwent breast cancer surgery and CancerSCANTM in 2015. The median follow-up period was 26 months (range, 1–46), and the average distant metastasis-free interval was 21 months (range, 1–46). When a threshold value of 0.5 was applied, the true-negative was 81, and the true-positive was two among the 83 patients. No false negative or false positive results were observed. The validation accuracy was 1.000.
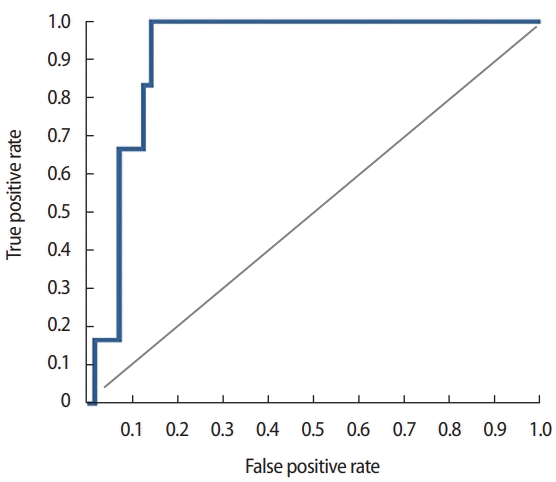
Receiver operating characteristic (ROC) curve. The area under the ROC curve (AUC) was 0.917.
Clinical application
The Azure ML platform provides a function for setting up web services (http://docs.microsoft.com/en-us/azure/machine-learning/studio/consume-web-services). After the Azure ML predictive model was used as a web service, we utilized a representational state-transfer application programming interface to send data and obtain real-time predictions. For example, when data (0 or 1) were inputted according to each variable, excluding the final value (distant metastasis), an external application communicated with a machine-learning workflow scoring model in real-time, enabling the predicted value to be calculated in a few seconds (Figure 3).
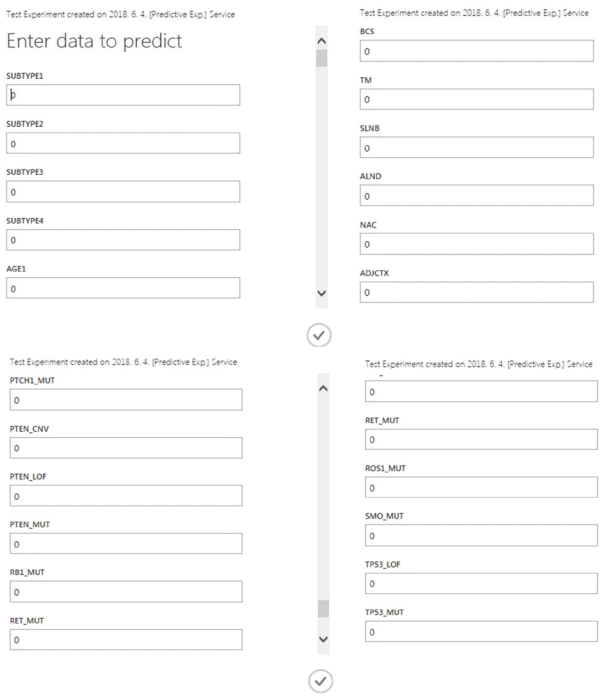
Illustration of web service usage for our predictive tool. BCS= breastconserving surgery; TM= total mastectomy; SLNB=sentinel lymph node biopsy; ALND=axillary lymph node dissection; NAC=neoadjuvant chemotherapy; ADJCTX=adjuvant chemotherapy; MUT=mutation; CNV=copy number variation; LOF=loss-of-function.
DISCUSSION
Distant metastasis from primary breast cancer is potentially lethal and has a complex mechanism. A commonly accepted theory is that as cancer grows, cells within the tumor acquire the capability to spread, survive, and flourish within the regional lymph nodes and other distant sites [7]. In addition, models of metastatic spread describe the complex interaction between seed and soil factors involving tumor intravasation, circulation, extravasation, proliferation, angiogenesis [21], and the microenvironment of the target tissue [22,23]. Owing to the heterogeneous nature of breast cancer metastasis, it is difficult to define a cure for this disease and assess the risk factors for metastasis [24].
A previous study identified the presence of lymph node metastasis, large primary tumor, and loss of histopathological differentiation (grade) as breast cancer prognostic markers [2]. A study of nomograms to predict metastasis-free survival used clinical findings, such as estrogen receptor (ER) status, histological grade, age, and chemotherapy cycles; however, the concordance index was 0.72 [25].
As gene expression signatures of human primary breast tumors enable more accurate predictions than prognostic factors, patients are destined to relapse and ultimately die due to metastatic breast cancer [2]. Several studies have sought to predict distant metastasis using gene expression. Cheng et al. [9] developed an 18-gene classifier to estimate distant metastasis risk. The 18-gene scoring system classified patients into low- and high-risk groups. Based on external validation, the 5-year probability of freedom from distant metastasis was 89.5% for low-risk patients and 73.6% for high-risk patients (p = 0.003) [9]. Wang et al. [8] used gene expression profiles to predict distant metastases. These researchers identified a 76-gene signature using an RNA microarray from 286 patients, which showed 93% sensitivity and 48% specificity in 171 independent testing sets [8]. Zemmour et al. [26] conducted DNA microarray studies that identified gene expression signatures for predicting metastatic relapse in early breast cancer. Using only six genes, the Cox Boost classifier predicted the 4-year status of metastatic disease with 93% sensitivity [26].
We developed a new predictive tool for distant metastasis using the clinical characteristics and gene profiles of 27 genes and 34 occasional results (mutation, loss-of-function, or copy number variation). Our study is valuable as it consisted of clinical findings and gene profiles and was conducted using ML. The Azure ML platform used in this study offers several advantages: real-time analysis can be performed in the clinical setting and the platform is free. An optional paid tool is available on the Azure ML platform; however, this study was adequately performed with the free-option tool. In addition, the Azure ML platform could be used to develop a suitable model for each hospital. The findings of a predictive study with large data collected at one center may not always be suitable for use by other institutions. This discrepancy may be due to differences in race or variable values. The accuracy of the predictive tool was dependent on accurate variable information (e.g., histological grading or ER, progesterone receptor, and HER2 immunohistochemical results). These factors were measured according to the official international standards. However, minimal differences may exist between centers and individual patients. Our predictive model can incorporate data from other centers or hospitals and provide proper results for each center; thus, any disparity among centers or hospitals could be reduced.
We developed an additional predictive tool for distant metastatic sites based on the data from 59 patients in the distant metastasis group. A Multi-class Neural Network was used for the analysis, and the overall accuracy was 0.86 (Supplementary Figure 2). When a prospective internal validation was conducted using 83 patients who underwent breast cancer surgery and CancerSCANTM in 2015, this tool did not predict accurate sites. Among the 83 patients, one had lung metastasis and one had multiple (bone and liver) metastases, where the predictive sites were the liver and bone, respectively. This result was due to the small number of patients with distant metastasis. In addition, the follow-up period was shorter than that of the modeling and test groups.
Our study had several limitations. First, our gene data were collected from tissues during surgery, regardless of whether patients received or did not receive neoadjuvant therapy. Therefore, it was indistinguishable from gene transformation induced by chemotherapy. Second, the number of patients enrolled in this study was small. Third, only an internal validation was performed. Fourth, we did not include time as a factor in our analysis. Therefore, our model predicts the presence or absence of metastasis but not when metastasis occurs.
Our predictive model is a useful and easy-to-access tool for identifying patients with distant metastases. Our model presents a way for each institution to achieve optimal results using its variables and also helps clinical decision for metastasis work up during follow up period. Further evaluations with a larger patient population will improve the reliability of this model.
Notes
The authors declare that they have no competing interests.
Acknowledgements
The authors thank Dr. Sung Wook Seo for providing advice on machine learning analysis.
SUPPLEMENTARY MATERIAL
Supplementary Figure 1.
(A) List of genes in order of importance score estimated by gradient boosting: A total of 243 features (alterations, including loss-offunction, mutation, and copy number variation in 81 genes) were used as input features. The target value was alive or dead at the 5-year mark. The genes were sorted in descending order of importance. (B) Selection for the optimal number of genes: Bootstrap resampling (n=100) was performed by sequentially reducing the less important genetic features, where the training sets (85%) and their corresponding test sets (15%) were resampled 100 times. The performance of each model was evaluated using the AUC values. The AUC was sequentially reduced by reducing the number of features. The AUC values of the 27 important genes (0.81; 95%CI: 0.76–0.85) were not significantly different from those with a higher number of genes. However, the AUC values of 26 important genes (0.724; 95% CI: 0.80–0.66) were significantly lower than those of the 27 important genes (p=0.000).
Supplementary Figure 2.
Prediction results for distant metastatic sites based on the data of 59 patients in the distant metastasis group.
Class 1: Bone; Class 2: Liver; Class 3: Lung; Class 4: Brain; Class 5: Others; Class 6: Multiple sites.